Do not delete.
Adeno-associated viruses (AAVs) serve as vectors for delivering therapeutic genes into targeted cells and tissues in a patient’s body. These viruses are non-pathogenic to humans. AAVs consist of a protein coat, known as the capsid, which encapsulates genetic material, such as DNA. Various strains or serotypes of AAV can target different cells and organs in the body. PaVe-GT is developing four gene therapies using AAV serotypes 8 and 9 to deliver therapeutic genes. In AAV-mediated gene therapy, most of the viral genes in the AAV particle are replaced with the therapeutic human gene.
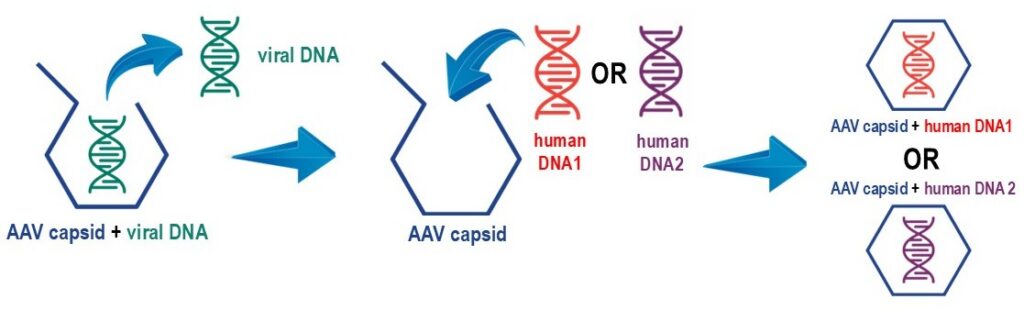
Different AAV-mediated gene therapies can be produced by introducing various therapeutic genes in the AAV capsid. This is why AAV is sometimes called a “platform” therapeutic.
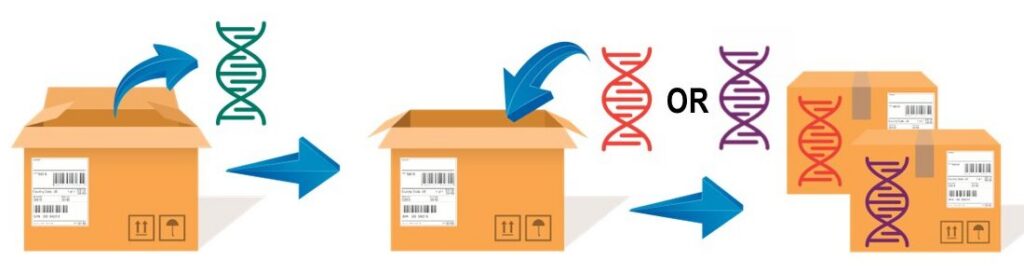
In principle, you can think of AAV as a shipping box. In AAV-mediated gene therapy, the AAV capsid acts like a shipping box, delivering the therapeutic gene to its target. If the gene fits in the capsid, is packed correctly, and the address is serviceable, it will reach its destination. The gene only becomes relevant once delivered. This method allows different genes to be inserted into the AAV capsid, making AAV and similar vectors valuable for treating various genetic diseases.
In practice, the process is much more complicated and involves three phases:
After purification, the product undergoes a series of tests to ensure it is pure and of desired quality for safe administration to humans.
Reference
Wang, J.H., et al. (2024) Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 9(1):78.
In addition to the information above, please visit our Other Resources page for patient-friendly explanations of gene therapy.
Of the 10,000 known rare diseases, 80% are of genetic origin. Diseases with a single gene defect could theoretically be treated with gene replacement therapy using AAV vectors. The potential to develop gene therapies for more diseases is growing, however, developing new gene therapies is a lengthy, costly, and complex process. In addition to the scientific challenges, operational and financial barriers exist, especially for conditions affecting very few patients. PaVe-GT is part of NCATS’ efforts to streamline gene therapy development for rare diseases by addressing scientific and operational hurdles during preclinical to clinical translation. The program aims to “pave the way” toward a public model for developing AAV gene therapies and conducting first-in-human clinical trials for small patient groups, unlikely to attract commercial interest. Learnings and resources from PaVe-GT can contribute to the gene therapy field at large by providing a blueprint for future projects.
The PaVe-GT program aims to streamline the start of gene therapy clinical trials for four rare diseases: two congenital myasthenic syndromes and two organic acidemias. The organic acidemias are PCCA-related propionic acidemia and cobalamin type B methylmalonic acidemia (MMAB gene defects). The congenital myasthenic syndromes are due to mutations in DOK7 or COLQ genes. To boost PaVe-GT’s success, NIH will conduct the trials with its investigators and resources at the NIH Clinical Center. Since all four trials will occur at NIH, project learnings and results can be made publicly accessible.
The selection of these diseases for PaVe-GT was based on the following reasons:
We have chosen specific diseases for this pilot program and cannot add more at this time. However, PaVe-GT’s results will aid many future gene therapy trials as we will share insights on the NIH PaVe-GT website for everyone’s benefit.
The disease summaries are provided below. For a patient-friendly description, please visit the Genetic and Rare Disease Information Center website propionic acidemia or methylmalonyl-Coenzyme A mutase deficiency.
Organic acidemias (OAs) are inherited metabolic disorders which disrupt branched-chain amino acid, odd-chain fatty acid and/or cholesterol metabolism and cause an accumulation of organic acids in the blood. Methylmalonic acidemia and propionic acidemia are common types of OAs. Newborns with OAs often experience a “crisis,” such as severe metabolic acidosis and high ammonia levels in the blood, which can be life-threatening and require intensive care or hemodialysis. During infancy and childhood, symptoms may include lethargy, vomiting, poor feeding, failure to thrive, low muscle tone, seizures, developmental delays, and severe illness triggered by minor stressors like colds. Metabolic decompensation can occur during periods of increased catabolism, such as illness, stress, trauma, surgery, or prolonged fasting. In the U.S., most OAs are detected through newborn screening. Treatment typically involves dietary restrictions, special formulas, carnitine supplements, and vitamin B12 injections for some MMA patients. Severely affected individuals might undergo liver transplants to boost hepatic enzyme activity. Overall, long-term clinical outcomes for OAs are generally poor, highlighting the need for new treatment options.
Propionic acidemia:
Propionic acidemia (PA) is a rare metabolic disorder caused by a deficiency in the propionyl-CoA carboxylase (PCC) enzyme. This enzyme consists of alpha and beta subunits encoded by the PCCA and PCCB genes, with mutations found equally in both. PA commonly presents in newborns with a potentially fatal metabolic crisis. Treatment includes dietary restriction of branched-chain amino acids, carnitine supplementation, and careful management of infections and stress. Despite these efforts, patients may experience severe complications, such as metabolic decompensations, hyperammonemia, pancreatitis, strokes, poor growth, and cytopenias. Elective liver transplantation has been used to restore PCC activity and improve symptoms. Additionally, AAV gene therapy shows promise based on preclinical studies in murine models.
Methylmalonic acidemia:
Isolated methylmalonic acidemia (MMA) comprises a group of varied metabolic disorders resulting from a complete or partial deficiency of the mitochondrial enzyme, methylmalonyl-CoA mutase (MMUT). One of the rarest forms, cobalamin B type MMA, is caused by mutations in the MMAB gene. This type is inherited in an autosomal recessive manner and can be identified through newborn screening and confirmed via genetic testing. Symptoms in patients often appear during infancy or the first year of life and include acid-base imbalance, high ammonia levels, lethargy, vomiting, dehydration, hypotonia, and delayed development. Long-term issues may include kidney disease, pancreatitis, cytopenias, and neurological damage due to metabolic strokes of the basal ganglia. Current treatments involve protein dietary restriction, vitamin B12 injections, carnitine, and intermittent antibiotics. However, there is no cure, and patients may still face significant medical challenges despite these interventions. Preclinical AAV gene therapy has successfully treated mouse models of MMA related to MMUT genes and is expected to be similarly effective for cobalamin B type MMA.
References:
The disease summaries are provided below. For a patient-friendly description, please visit the Genetic and Rare Disease Information Center website: congenital myasthenic syndromes.
Congenital myasthenic syndromes (CMS) are a group of inherited disorders in which there is impairment of the highly specialized neuromuscular junction (NMJ). To date, 32 types of CMS have been described. Defects in all areas of the NMJ — including the nerve and muscle cells — have been identified as molecular causes for CMS, and most types are inherited in an autosomal recessive pattern with early onset. The four most common recessively inherited forms of CMS result from mutations in the DOK7, COLQ, ACHE or RAPSN genes. While symptoms vary depending on the underlying cause of the disease, they may include muscle and respiratory weakness, drooping eyelids, delayed motor development, fatigue and contractures. These NMJ disorders are not primarily degenerative, and rebuilding the NMJ will restore function to the neuromuscular unit. The NMJ target is of further interest for broader applications, including amyotrophic lateral sclerosis (ALS), sarcopenia and myasthenia gravis. The two forms of CMS chosen for the PaVe-GT initiative (Dok7 and ColQ deficiencies) account for ~ 20% of all CMS cases.
COLQ-related CMS:
Collagen Q, a specific nonfibrillar collagen encoded by the COLQ gene, is essential for anchoring the neurotransmitter acetylcholinesterase in the NMJ. In individuals with COLQ-related CMS, acetylcholinesterase is not anchored in the synaptic cleft, resulting in altered NMJ signaling. Over 30 mutations in COLQ have been identified as causative for CMS. Similar to DOK7-related CMS, initial symptoms typically appear during the neonatal period or early in childhood and include muscle hypotonia, fatigue, respiratory insufficiency, and delayed motor development. Current treatment options, such as salbutamol and ephedrine, yield limited responses.
DOK7-related CMS:
The DOK7 gene encodes the Dok7 protein, crucial for NMJ function. Patients with DOK7 deficiency often have difficulty walking at disease onset and may experience facial, jaw, and neck weaknesses. Unlike other CMS types, these patients might not show significant fluctuations in weakness or fatigability, possibly leading to a misdiagnosis of fixed myopathy. Respiratory function can progressively decline, but eye movements typically remain unaffected. There’s no clear link between clinical symptoms and different mutations in the gene. Treatments include pyridostigmine, ephedrine (β2-adrenergic agonist), and 3,4-diaminopyridine, though responses vary.
References:
There are three natural history studies being conducted at the NIH Clinical Center that are directly relevant to the diseases being targeted by PaVe-GT:
Patients and caregivers looking for information about these diseases are welcome to contact the NIH Genetic and Rare Diseases Information Center.