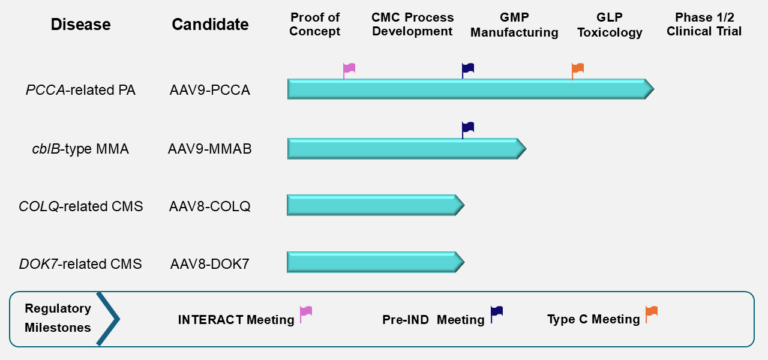
The figure below summarizes the current stage of preclinical development for the four PaVe-GT lead candidates. Although progress is represented linearly, preclinical development activities overlap and are interdependent. The PCCA program, with AAV9-hPCCA, is farthest along, and we are currently preparing for IND submission. The lead candidates AAV9-hPCCA and AAV9-MMAB have also been awarded the Orphan Drug and Rare Pediatric Disease Designations from the FDA, which offer several incentives to drug developers.
The AAV9 serotype is used for the PCCA and MMAB candidates, and the AAV8 serotype is used for the COLQ and DOK7 candidates. The PCCA and COLQ candidates are engineered as single-stranded AAV vectors, while the MMAB and DOK7 candidates use a self-complementary design.
Abbreviations: cblB, cobalamin B; CMC, Chemistry, Manufacturing, and Controls; CMS, Congenital Myasthenic Syndrome; COLQ, Collagen Q; DOK7, Downstream of Tyrosine Kinase 7; GLP, Good Laboratory Practices; GMP, Good Manufacturing Practices; IND, Investigational New Drug; INTERACT, INitial Targeted Engagement for Regulatory Advice for CBER/CDER producTs; MMA, Methylmalonic Acidemia; MMAB, Metabolism of Cobalamin Associated B; PA, Propionic Acidemia; PCCA, Propionyl-Coenzyme A Carboxylase Alpha subunit